ASE Basic¶
Pythonで原子シミュレーションを進めていく際に便利なOSSライブラリとして、Atomic Simulation Environment: ASEがあります。 本節ではASEの基本的な使い方を紹介していきます。
Documentation: https://wiki.fysik.dtu.dk/ase/
gitlab: https://gitlab.com/ase/ase
Atomsについて¶
ASEでは原子が複数個集まって作られる系を Atoms クラスで表現します。
Atomsクラスは前節で説明した、原子シミュレーションに必要な構造を表現するための以下のようなattribute(変数)を保持しています。
各原子の元素種類
座標値
速度(運動量)
セル
周期境界条件など
Atomsの作成方法: 原子種・座標を直接指定¶
Atomsを作るための一番原始的な方法として、原子種とその座標値を直接指定する方法があります。 以下は、1つめのHがxyz座標値 [0, 0, 0]、2つめのHがxyz座標値 [1.0, 0, 0] に存在する水素分子H2を作る例です。
[1]:
from ase import Atoms
atoms = Atoms("H2", [[0, 0, 0], [1.0, 0, 0]])
原子の可視化を行うこともできます。 ここでは、ASEのview 関数を用い、nglviewerというライブラリを用いて可視化を行っています。
nglviewer を用いると、3次元の原子構造を表示しながら、マウスでインタラクティブに操作することもできます。
可視化についての詳細はAppendix_1_visualization.ipynbをご参照ください。
[2]:
from ase.visualize import view
from pfcc_extras.visualize.view import view_ngl
#view(atoms, viewer="ngl")
view_ngl(atoms, representations=["ball+stick"], w=400, h=300)
[2]:
以下のように、ASEを用いてpng静止画を描画することも可能です。
[3]:
from ase.io import write
from IPython.display import Image
write("output/H2.png", atoms, rotation="0x,0y,0z", scale=100)
Image(url='output/H2.png', width=150)
[3]:

以下は上記のプログラムを1行でできるようにした便利関数です。今後はこれを用います。
[4]:
from pfcc_extras.visualize.ase import view_ase_atoms
view_ase_atoms(atoms, figsize=(4, 4), title="H2 atoms", scale=100)
元素記号 symbolsの代わりに、原子番号をnumbers として指定することも可能です。
[5]:
co_atoms = Atoms(numbers=[6, 8], positions=[[0, 0, 0], [1.0, 0, 0]])
view_ngl(co_atoms, representations=["ball+stick"], w=400, h=300)
[5]:
[6]:
view_ase_atoms(co_atoms, figsize=(4, 4), title="CO atoms", scale=100)
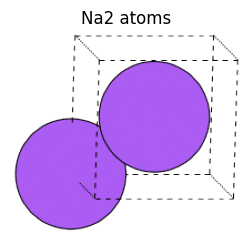
周期境界条件を含む系を定義したい場合は、cell に周期情報を指定し、pbc にa軸b軸c軸それぞれに対して周期境界条件を適用するかどうかのON/OFFを指定することができます。
[7]:
from ase import Atoms
na2_atoms = Atoms(
symbols="Na2",
positions=[[0, 0, 0], [2.115, 2.115, 2.115]],
cell=[4.23, 4.23, 4.23],
pbc=[True, True, True]
)
[8]:
view_ngl(na2_atoms, w=400, h=300)
[8]:
[9]:
view_ase_atoms(na2_atoms, rotation="10x,10y,0z", figsize=(3, 3), title="Na2 atoms", scale=30)
Atomsの持つattribute, method¶

atoms はその系の情報を保持しており、Attribute や get_XXX 関数を通して参照することができます。
Attributeとは Atomsクラスの持つ変数のことで、 atoms.xxx と直接参照することが出来るものもあります。 関数を通してアクセスできる情報もあります。
以下は主要なAttributeや関数の返り値(numpy array としてのshapeがどの様になっているか)をまとめたものです。
Attribute
関数
get_masses(): 各原子の質量を返す。get_momenta(): 各原子に設定されている運動量を返す。len(atoms): Atoms全体の原子数Nを返す。
[10]:
print(f"symbols : {atoms.symbols}")
print(f"positions: {atoms.positions}")
print(f"cell : {atoms.cell}")
print(f"pbc : {atoms.pbc}")
# "Atomic numbers" and "Mass" can be automatically calculated from "Symbol"
print(f"numbers : {atoms.numbers}")
print(f"massess : {atoms.get_masses()}")
print(f"momenta : {atoms.get_momenta()}")
print(f"Numbers of atoms : {len(atoms)}")
symbols : H2
positions: [[0. 0. 0.]
[1. 0. 0.]]
cell : Cell([0.0, 0.0, 0.0])
pbc : [False False False]
numbers : [1 1]
massess : [1.008 1.008]
momenta : [[0. 0. 0.]
[0. 0. 0.]]
Numbers of atoms : 2
単位系¶
ASEでは、エネルギーの単位はeV (約 \(1.602 \times 10^{-19}\) J)、座標系の単位はÅ (\(1 \times 10^{-10}\) m)で扱われる事が多いです。 力や応力の単位はこれらの複合単位で、力についてはeV/Å、応力についてはeV/Å2です。chargeの単位は電荷素量です。
[Tips] Jupyter 上で各クラス・関数のより詳細な情報を知りたい場合、 ? を後ろにつけることでドキュメントの表示ができます。
[11]:
Atoms?
Init signature:
Atoms(
symbols=None,
positions=None,
numbers=None,
tags=None,
momenta=None,
masses=None,
magmoms=None,
charges=None,
scaled_positions=None,
cell=None,
pbc=None,
celldisp=None,
constraint=None,
calculator=None,
info=None,
velocities=None,
)
Docstring:
Atoms object.
The Atoms object can represent an isolated molecule, or a
periodically repeated structure. It has a unit cell and
there may be periodic boundary conditions along any of the three
unit cell axes.
Information about the atoms (atomic numbers and position) is
stored in ndarrays. Optionally, there can be information about
tags, momenta, masses, magnetic moments and charges.
In order to calculate energies, forces and stresses, a calculator
object has to attached to the atoms object.
Parameters
----------
symbols : str | list[str] | list[Atom]
Chemical formula, a list of chemical symbols, or list of
:class:`~ase.Atom` objects (mutually exclusive with ``numbers``).
- ``'H2O'``
- ``'COPt12'``
- ``['H', 'H', 'O']``
- ``[Atom('Ne', (x, y, z)), ...]``
positions : list[tuple[float, float, float]]
Atomic positions in Cartesian coordinates
(mutually exclusive with ``scaled_positions``).
Anything that can be converted to an ndarray of shape (n, 3) works:
[(x0, y0, z0), (x1, y1, z1), ...].
scaled_positions : list[tuple[float, float, float]]
Atomic positions in units of the unit cell
(mutually exclusive with ``positions``).
numbers : list[int]
Atomic numbers (mutually exclusive with ``symbols``).
tags : list[int]
Special purpose tags.
momenta : list[tuple[float, float, float]]
Momenta for all atoms in Cartesian coordinates
(mutually exclusive with ``velocities``).
velocities : list[tuple[float, float, float]]
Velocities for all atoms in Cartesian coordinates
(mutually exclusive with ``momenta``).
masses : list[float]
Atomic masses in atomic units.
magmoms : list[float] | list[tuple[float, float, float]]
Magnetic moments. Can be either a single value for each atom
for collinear calculations or three numbers for each atom for
non-collinear calculations.
charges : list[float]
Initial atomic charges.
cell : 3x3 matrix or length 3 or 6 vector, default: (0, 0, 0)
Unit cell vectors. Can also be given as just three
numbers for orthorhombic cells, or 6 numbers, where
first three are lengths of unit cell vectors, and the
other three are angles between them (in degrees), in following order:
[len(a), len(b), len(c), angle(b,c), angle(a,c), angle(a,b)].
First vector will lie in x-direction, second in xy-plane,
and the third one in z-positive subspace.
celldisp : tuple[float, float, float], default: (0, 0, 0)
Unit cell displacement vector. To visualize a displaced cell
around the center of mass of a Systems of atoms.
pbc : bool | tuple[bool, bool, bool], default: False
Periodic boundary conditions flags.
- ``True``
- ``False``
- ``0``
- ``1``
- ``(1, 1, 0)``
- ``(True, False, False)``
constraint : constraint object(s)
One or more ASE constraints applied during structure optimization.
calculator : calculator object
ASE calculator to obtain energies and atomic forces.
info : dict | None, default: None
Dictionary with additional information about the system.
The following keys may be used by ASE:
- spacegroup: :class:`~ase.spacegroup.Spacegroup` instance
- unit_cell: 'conventional' | 'primitive' | int | 3 ints
- adsorbate_info: Information about special adsorption sites
Items in the info attribute survives copy and slicing and can
be stored in and retrieved from trajectory files given that the
key is a string, the value is JSON-compatible and, if the value is a
user-defined object, its base class is importable. One should
not make any assumptions about the existence of keys.
Examples
--------
>>> from ase import Atom
N2 molecule (These three are equivalent):
>>> d = 1.104 # N2 bondlength
>>> a = Atoms('N2', [(0, 0, 0), (0, 0, d)])
>>> a = Atoms(numbers=[7, 7], positions=[(0, 0, 0), (0, 0, d)])
>>> a = Atoms([Atom('N', (0, 0, 0)), Atom('N', (0, 0, d))])
FCC gold:
>>> a = 4.05 # Gold lattice constant
>>> b = a / 2
>>> fcc = Atoms('Au',
... cell=[(0, b, b), (b, 0, b), (b, b, 0)],
... pbc=True)
Hydrogen wire:
>>> d = 0.9 # H-H distance
>>> h = Atoms('H', positions=[(0, 0, 0)],
... cell=(d, 0, 0),
... pbc=(1, 0, 0))
File: ~/.py311/lib/python3.11/site-packages/ase/atoms.py
Type: type
Subclasses: MSONAtoms, Cluster, Lattice