ASE Structure¶
構造生成の方法¶
前節で説明した、直接 Atoms に座標値を指定する方法は、1つ1つの原子位置を細かく設定したい場合には良いですが、 通常使う場合には各原子の座標値を明示的に調べて設定する必要があり、大変です。
ASEでは、いろいろな構造を作成するための関数がすでに定義されており、これらを用いると簡単に構造を作成することができます。
[1]:
from ase import Atoms
from ase.visualize import view
from pfcc_extras.visualize.view import view_ngl
from pfcc_extras.visualize.ase import view_ase_atoms
ASE molecule¶
molecule methodはASEで登録済みの有機分子を作成することができます。 登録済みの名前リストは以下のように見ることができます。
[2]:
from ase.build import molecule
from ase.collections import g2
print(f"Available molecule:", len(g2.names), g2.names)
Available molecule: 162 ['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF', 'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2', 'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3', 'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane', 'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2', 'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl', 'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN', 'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO', 'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2', 'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C', 'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S', 'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d', 'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O', 'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF', 'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4', 'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O', 'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO', 'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl', 'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2']
以下のように文字列を指定するだけで atomsの作成ができます。
[3]:
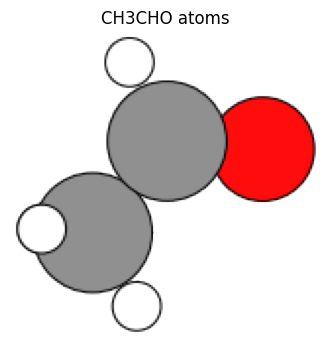
ch3cho_atoms = molecule("CH3CHO")
view_ngl(ch3cho_atoms, representations=["ball+stick"], w=400, h=300)
[3]:
[4]:
view_ase_atoms(ch3cho_atoms, rotation="0x,0y,0z", figsize=(4, 4), title="CH3CHO atoms", scale=40)
ASE bulk¶
bulk methodは結晶構造を簡単に作ることができます。
以下のように、結晶を特徴づける値を指定することでその結晶構造が生成されます。
name: 原子種crystalstructure: “sc”, “fcc”, “bcc” などの結晶構造を指定a, b, c, alpha, covera: Cellの形や大きさを指定cubic: Cellの形をCubic unit cell にするか否か
[5]:
from ase.build import bulk
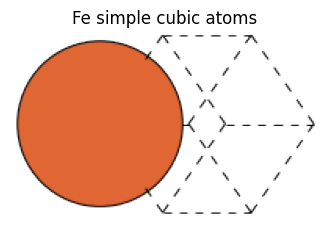
fe_sc_atoms = bulk(name="Fe", crystalstructure="sc", a=2.0)
fe_sc_atoms
[5]:
Atoms(symbols='Fe', pbc=True, cell=[2.0, 2.0, 2.0])
次の可視化では1原子しか表示されていません。ズームアウトして、全体を見てみると、黄色い枠でセルが表示されていることがわかります。
[6]:
view_ngl(fe_sc_atoms, w=400, h=300)
[6]:
詳しくは後述しますが、セルと周期境界条件(pbc)が与えられた場合、上記の構造は周期境界条件に従って無限に続いているような構造を考えます。 つまり表示上は1原子しか表示されていませんが、以下の構造のように規則正しく構造が続いて配置されている結晶構造を表しています。
[7]:
view_ngl(fe_sc_atoms * (6, 6, 6), w=400, h=300)
[7]:
[8]:
view_ase_atoms(fe_sc_atoms, rotation="45x,45y,0z", figsize=(4, 4), title="Fe simple cubic atoms", scale=40)
crystalstructureやCell size a などを指定しなかった場合、ASEが元素種から自動で最適なものを決定します。
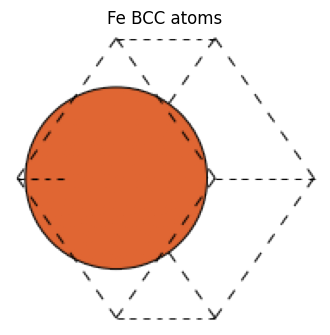
例えば、Feの場合 a=2.87のBCC構造となります。
[9]:
fe_atoms = bulk("Fe")
fe_atoms
[9]:
Atoms(symbols='Fe', pbc=True, cell=[[-1.435, 1.435, 1.435], [1.435, -1.435, 1.435], [1.435, 1.435, -1.435]], initial_magmoms=...)
[10]:
view_ngl(fe_atoms, w=400, h=300)
[10]:
[11]:
view_ase_atoms(fe_atoms, rotation="45x,0y,0z", figsize=(4, 4), title="Fe BCC atoms", scale=40)
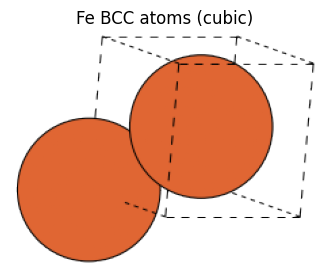
上記のように、BCCやFCC構造の場合、原子を1つのみ含む単位格子(unit cell)である基本単位胞(primitive cell)のCellは立方体ではありませんが、単位格子を拡張してとることにより立方体の形にすることも可能です。 cubic=True の引数を設定することで立方体の格子を持つ Atomsが生成できます。
これらのPrimitive Cellと立方体のCellをもつUnit Cellの違いは、以下の文献が参考になります。
[12]:
fe_cubic_atoms = bulk("Fe", cubic=True)
fe_cubic_atoms
[12]:
Atoms(symbols='Fe2', pbc=True, cell=[2.87, 2.87, 2.87], initial_magmoms=...)
[13]:
view_ngl(fe_cubic_atoms, w=400, h=300)
[13]:
[14]:
view_ase_atoms(fe_cubic_atoms, rotation="10x,30y,0z", figsize=(4, 4), title="Fe BCC atoms (cubic)", scale=40)
repeat関数
周期境界条件を持つ構造は repeat 関数を使うことで、周期を繰り返しSuper cellとすることができます。
以下の例では a軸方向に2倍、 b軸方向に3倍、 c軸方向に4倍、の大きさのCellの原子構造を作成しています。
[15]:
fe234_atoms = fe_atoms.repeat((2, 3, 4))
view_ngl(fe234_atoms, w=400, h=300)
[15]:
repeat関数の代わりに、掛け算で指定することもできます。
[16]:
fe234_atoms = fe_atoms * (2, 3, 4)
view_ngl(fe234_atoms, w=400, h=300)
[16]:
[17]:
view_ase_atoms(fe234_atoms, rotation="45x,0y,0z", figsize=(5, 5), title="Fe BCC atoms (2x2x2)", scale=30)
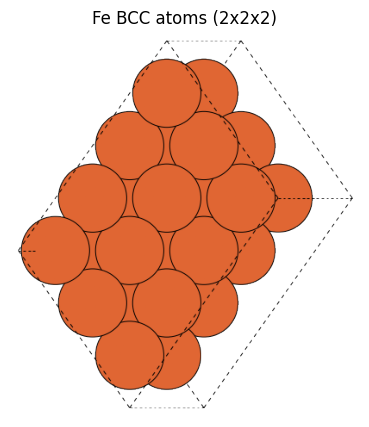
このように周期境界条件に従って無限に続くことを考えると、cubicを指定せずにprimitive cellで表現されたFe構造と、cubic=Trueとしてセルを立方体にとったUnit cellのFe構造が同じ構造を表していることを確認することができます。
また bulk methodは2元素からなる構造なども生成することができます。
[18]:
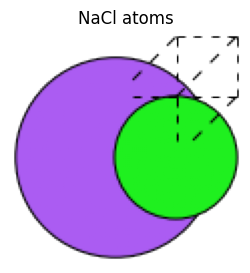
nacl_atoms = bulk("NaCl", crystalstructure="rocksalt", a=2.0)
view_ngl(nacl_atoms * (3, 3, 3), w=400, h=300)
[18]:
[19]:
view_ase_atoms(nacl_atoms, rotation="0x,0y,0z", figsize=(3, 3), title="NaCl atoms", scale=30)
ASE 表面構造¶
触媒との反応を扱う場合などは、金属結晶の表面に対して反応物がどのように振る舞うかを見る形でモデリングを行う場合があります。 結晶の表面をミラー指数で指定することができます。
ASEでは主要な結晶構造に対する、各表面を指定されたミラー指数で切り出されたものを作成することができます。表面はz軸方向に作られます。 このような表面がでている構造を Slab構造と呼びます。
Slab構造の作成時には以下のようなパラメータを指定します。
symbol: 元素種を指定size: x, y, z軸それぞれの方向への原子数を指定vacuum: z軸上下に作成する真空層の厚さを指定
[20]:
from ase.build import (
fcc100, fcc110, fcc111, fcc211, fcc111_root,
bcc100, bcc110, bcc111, bcc111_root,
hcp0001, hcp10m10, hcp0001_root,
diamond100, diamond111
)
# fcc: fcc100(), fcc110(), fcc111(), fcc211(), fcc111_root()
# bcc: bcc100(), bcc110(), bcc111() * - bcc111_root()
# hcp: hcp0001(), hcp10m10(), hcp0001_root()
# diamond: diamond100(), diamond111()
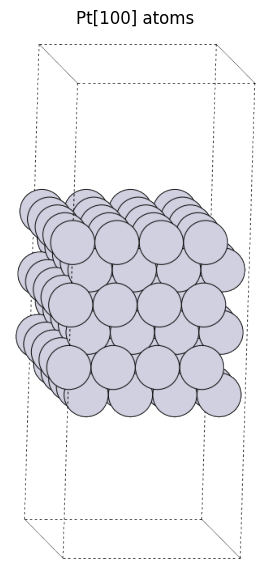
pt100_atoms = fcc100("Pt", size=(4, 5, 6), vacuum=10.0)
[21]:
view_ase_atoms(pt100_atoms, rotation="-80x,10y,0z", figsize=(7.0, 7.0), title="Pt[100] atoms", scale=20)
[22]:
fcc100?
Signature: fcc100(symbol, size, a=None, vacuum=None, orthogonal=True, periodic=False)
Docstring:
FCC(100) surface.
Supported special adsorption sites: 'ontop', 'bridge', 'hollow'.
File: ~/.py311/lib/python3.11/site-packages/ase/build/surface.py
Type: function
しばしば表面の片方に注目して解析を行いますが、その場合であっても、反対側にももう1つの表面が出ることに注意してください。通常は反対側の表面の影響を受けないよう、ある程度の厚みをもたせて(この例では6層)解析を行います。
SMILESからの生成¶
有機分子を表す際には、SMILES記法を用いて、特定の分子を表すことも多いです。 例えばエチレンは “C=C” で表すことができます。
以下の関数は、内部で有機分子を扱うライブラリである RDKit を使用して、SMILES構造からのAtoms生成を行っています。
[23]:
from pfcc_extras.structure.ase_rdkit_converter import smiles_to_atoms, atoms_to_smiles
atoms = smiles_to_atoms("C=C")
view_ngl(atoms, representations=["ball+stick"], w=400, h=300)
[23]:
atomsからSMILESに戻す事もできます。(Experimentalな機能で、正しく動かない場合もありますのでご注意ください)
[24]:
atoms_to_smiles(atoms)
[24]:
'C=C'
ファイルから読み込む¶
ASEでは、 xyz, cif, VASPのPOSCARファイルなどなど、様々な原子構造を記述するファイルの書き込み・読み込みに対応しています。 対応しているファイルの一覧は以下のドキュメントを参照ください。
[25]:
from ase.io import read, write
# write to file
write("output/pt100.xyz", pt100_atoms)
/home/jovyan/.py311/lib/python3.11/site-packages/ase/io/extxyz.py:318: UserWarning: Skipping unhashable information adsorbate_info
warnings.warn('Skipping unhashable information '
[26]:
# read from file
read("output/pt100.xyz")
[26]:
Atoms(symbols='Pt120', pbc=[True, True, False], cell=[11.087434329005065, 13.85929291125633, 29.8], tags=...)
外部データベースを使用する¶
Materials Project: https://materialsproject.org/
PubChem: https://pubchem.ncbi.nlm.nih.gov/
などといった世の中に存在する大規模データベースから、ファイルをダウンロードし構造を読み込んでくることも可能です。
これらファイルを使用する方法はMatlantis製品内のExampleに乗せておりますので、そちらをご覧ください。