Slab + mol energy¶
触媒上での反応などでは、
触媒表面上に有機分子が吸着
表面上で反応が進行
反応終了後、生成物が触媒表面上から脱離
といった過程を扱います。 反応物・生成物が触媒上で吸着・脱離をするため、吸着構造(表面構造上に分子が存在する構造)を扱う必要があります。
本節では、吸着構造に関連するエネルギーとして、吸着エネルギーの計算を取り扱います。
吸着エネルギー
[1]:
from ase import Atoms
from ase.build import bulk
from ase.constraints import ExpCellFilter, StrainFilter
from ase.optimize import LBFGS, FIRE
import pfp_api_client
from pfp_api_client.pfp.calculators.ase_calculator import ASECalculator
from pfp_api_client.pfp.estimator import Estimator, EstimatorCalcMode
print(f"pfp_api_client: {pfp_api_client.__version__}")
estimator = Estimator(calc_mode=EstimatorCalcMode.PBE, model_version="v8.0.0")
calculator = ASECalculator(estimator)
pfp_api_client: 1.23.1
吸着エネルギー¶
吸着エネルギー \(E_{\rm{adsorption}}\) は、ある分子が表面構造に吸着する前後のエネルギー差を表します。
\(E_{\rm{slab}}\)がSlab単体のエネルギー、\(E_{\rm{mol}}\)が分子単体のエネルギー、\(E_{\rm{slab+mol}}\)がSlab上に分子がついた吸着構造のエネルギーです。
CO on Co¶
[2]:
from ase.optimize import LBFGS
from ase.build import fcc111, hcp0001, molecule, add_adsorbate
from ase.constraints import ExpCellFilter, StrainFilter
def get_opt_energy(atoms, fmax=0.001, opt_mode: str = "normal"):
atoms.set_calculator(calculator)
if opt_mode == "scale":
opt1 = LBFGS(StrainFilter(atoms, mask=[1, 1, 1, 0, 0, 0]))
elif opt_mode == "all":
opt1 = LBFGS(ExpCellFilter(atoms))
else:
opt1 = LBFGS(atoms)
opt1.run(fmax=fmax)
return atoms.get_total_energy()
まずは適切なCo のセルサイズを求めます。
Bulkを用意して最適化することでセルサイズを求めます。
[3]:
bulk_atoms = bulk("Co")
bulk_atoms.cell
[3]:
Cell([[2.51, 0.0, 0.0], [-1.255, 2.173723763498941, 0.0], [0.0, 0.0, 4.07122]])
[4]:
bulk_atoms = bulk("Co")
bulk_atoms.calc = calculator
E_bulk = get_opt_energy(bulk_atoms, fmax=1e-4, opt_mode="scale")
E_bulk
Step Time Energy fmax
LBFGS: 0 05:00:53 -10.266314 0.917135
LBFGS: 1 05:00:53 -10.271767 0.190536
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
/tmp/ipykernel_3433/1344539460.py:9: FutureWarning: Import StrainFilter from ase.filters
opt1 = LBFGS(StrainFilter(atoms, mask=[1, 1, 1, 0, 0, 0]))
LBFGS: 2 05:00:53 -10.272057 0.065210
LBFGS: 3 05:00:54 -10.272109 0.007636
LBFGS: 4 05:00:54 -10.272111 0.000729
LBFGS: 5 05:00:54 -10.272112 0.000031
[4]:
-10.272111651260108
[5]:
bulk_atoms.cell
[5]:
Cell([[2.500496479864743, 0.0, 0.0], [-1.2502482399323716, 2.165493486666698, 0.0], [0.0, 0.0, 4.022806887309025]])
[6]:
# Set lattice parameter from bulk cell
a = bulk_atoms.cell[0,0]
c = bulk_atoms.cell[2,2]
a,c
[6]:
(2.500496479864743, 4.022806887309025)
ASE デフォルトのa = 2.51A からは少し値が変わっていることがわかります。このセルサイズを用いて以下、吸着構造を作成していきます。
[7]:
slab = hcp0001("Co", size=(4, 4, 4), a=a, c=c, vacuum=40.0, periodic=True)
slab.calc = calculator
[8]:
E_slab = get_opt_energy(slab, fmax=1e-4, opt_mode="normal")
Step Time Energy fmax
LBFGS: 0 05:00:54 -304.787566 0.465494
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
LBFGS: 1 05:00:54 -304.910147 0.370066
LBFGS: 2 05:00:54 -305.125230 0.054862
LBFGS: 3 05:00:54 -305.127507 0.037520
LBFGS: 4 05:00:54 -305.128856 0.039435
LBFGS: 5 05:00:54 -305.133149 0.032470
LBFGS: 6 05:00:54 -305.135449 0.017488
LBFGS: 7 05:00:54 -305.136081 0.006170
LBFGS: 8 05:00:54 -305.136162 0.000781
LBFGS: 9 05:00:54 -305.136161 0.000038
[9]:
mol = molecule("CO")
mol.calc = calculator
E_mol = get_opt_energy(mol, fmax=1e-4)
Step Time Energy fmax
LBFGS: 0 05:00:55 -11.443040 0.797117
LBFGS: 1 05:00:55 -11.431574 1.878720
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
LBFGS: 2 05:00:55 -11.445926 0.046015
LBFGS: 3 05:00:55 -11.445934 0.002568
LBFGS: 4 05:00:55 -11.445933 0.000001
吸着構造の作成¶
吸着構造は add_adsorbate 関数を用いることで、Slab上に分子を載せる事ができます。
[10]:
%%time
slab_mol = hcp0001("Co", size=(4, 4, 4), a=a, c=c, vacuum=40.0, periodic=True)
mol2 = molecule("CO")
add_adsorbate(slab_mol, mol2, 3.0, "bridge")
E_slab_mol = get_opt_energy(slab_mol, fmax=1e-2)
Step Time Energy fmax
LBFGS: 0 05:00:55 -317.298055 3.367191
LBFGS: 1 05:00:55 -317.432694 4.081281
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
LBFGS: 2 05:00:55 -317.699263 2.858175
LBFGS: 3 05:00:55 -317.973910 1.086529
LBFGS: 4 05:00:55 -317.996378 0.707769
LBFGS: 5 05:00:55 -318.071190 0.641089
LBFGS: 6 05:00:55 -318.081374 0.367003
LBFGS: 7 05:00:55 -318.087663 0.250177
LBFGS: 8 05:00:56 -318.107622 0.845603
LBFGS: 9 05:00:56 -318.115923 0.647254
LBFGS: 10 05:00:56 -318.121235 0.170493
LBFGS: 11 05:00:56 -318.123992 0.213722
LBFGS: 12 05:00:56 -318.127948 0.427355
LBFGS: 13 05:00:56 -318.130845 0.379397
LBFGS: 14 05:00:56 -318.132463 0.151676
LBFGS: 15 05:00:56 -318.133156 0.059567
LBFGS: 16 05:00:56 -318.133794 0.175820
LBFGS: 17 05:00:56 -318.134569 0.215512
LBFGS: 18 05:00:56 -318.135123 0.130339
LBFGS: 19 05:00:57 -318.135360 0.023292
LBFGS: 20 05:00:57 -318.135485 0.049542
LBFGS: 21 05:00:57 -318.135621 0.088976
LBFGS: 22 05:00:57 -318.135744 0.078587
LBFGS: 23 05:00:57 -318.135825 0.030664
LBFGS: 24 05:00:57 -318.135852 0.010856
LBFGS: 25 05:00:57 -318.135869 0.030200
LBFGS: 26 05:00:57 -318.135916 0.038714
LBFGS: 27 05:00:57 -318.135927 0.024988
LBFGS: 28 05:00:58 -318.135928 0.007101
CPU times: user 162 ms, sys: 11.7 ms, total: 174 ms
Wall time: 2.7 s
[11]:
E_adsorp = E_slab_mol - (E_slab + E_mol)
print(f"E_adsorp {E_adsorp}, E_slab_mol {E_slab_mol}, E_slab {E_slab}, E_mol {E_mol}")
E_adsorp -1.5538343284925418, E_slab_mol -318.13592752018087, E_slab -305.13616061153743, E_mol -11.445932580150906
吸着エネルギーとして-1.55 eVが得られました。
[12]:
from pfcc_extras.visualize.view import view_ngl
view_ngl(slab_mol)
[12]:
吸着サイトについて¶
add_adsorbate 関数の4番目に指定する position 引数として、座標値の代わりに吸着サイト名で指定することができます。
[13]:
%%time
slab_mol = hcp0001("Co", size=(4, 4, 4), a=a, c=c, vacuum=40.0, periodic=True)
mol2 = molecule("CO")
add_adsorbate(slab_mol, mol2, 3.0, "ontop")
E_slab_mol = get_opt_energy(slab_mol, fmax=1e-4)
Step Time Energy fmax
LBFGS: 0 05:01:04 -317.842476 2.898523
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
LBFGS: 1 05:01:04 -317.970380 3.594472
LBFGS: 2 05:01:04 -318.191337 1.677113
LBFGS: 3 05:01:05 -318.281429 0.675248
LBFGS: 4 05:01:05 -318.291021 0.402380
LBFGS: 5 05:01:05 -318.298278 0.037047
LBFGS: 6 05:01:05 -318.299164 0.060696
LBFGS: 7 05:01:05 -318.302288 0.184594
LBFGS: 8 05:01:05 -318.304052 0.160634
LBFGS: 9 05:01:05 -318.304860 0.053985
LBFGS: 10 05:01:05 -318.305023 0.024048
LBFGS: 11 05:01:05 -318.305066 0.019796
LBFGS: 12 05:01:05 -318.305139 0.025298
LBFGS: 13 05:01:05 -318.305123 0.013126
LBFGS: 14 05:01:05 -318.305191 0.004591
LBFGS: 15 05:01:05 -318.305180 0.004789
LBFGS: 16 05:01:05 -318.305185 0.009711
LBFGS: 17 05:01:06 -318.305189 0.010454
LBFGS: 18 05:01:06 -318.305212 0.005977
LBFGS: 19 05:01:06 -318.305179 0.001660
LBFGS: 20 05:01:06 -318.305149 0.002807
LBFGS: 21 05:01:06 -318.305150 0.003268
LBFGS: 22 05:01:06 -318.305208 0.002356
LBFGS: 23 05:01:06 -318.305210 0.001274
LBFGS: 24 05:01:06 -318.305172 0.001606
LBFGS: 25 05:01:06 -318.305206 0.002345
LBFGS: 26 05:01:06 -318.305210 0.002209
LBFGS: 27 05:01:06 -318.305215 0.001226
LBFGS: 28 05:01:06 -318.305168 0.000541
LBFGS: 29 05:01:06 -318.305219 0.000799
LBFGS: 30 05:01:07 -318.305208 0.000816
LBFGS: 31 05:01:07 -318.305170 0.000546
LBFGS: 32 05:01:07 -318.305226 0.000258
LBFGS: 33 05:01:07 -318.305159 0.000331
LBFGS: 34 05:01:07 -318.305154 0.000510
LBFGS: 35 05:01:07 -318.305206 0.000436
LBFGS: 36 05:01:07 -318.305153 0.000187
LBFGS: 37 05:01:07 -318.305170 0.000142
LBFGS: 38 05:01:07 -318.305219 0.000121
LBFGS: 39 05:01:07 -318.305208 0.000160
LBFGS: 40 05:01:07 -318.305180 0.000097
CPU times: user 217 ms, sys: 25.6 ms, total: 243 ms
Wall time: 3.08 s
この吸着サイトの名前は、 .info を表示することで “sites” 以下に定義されている値として確認ができます。
[14]:
slab_mol.info
[14]:
{'adsorbate_info': {'cell': array([[2.50049648, 0. ],
[1.25024824, 2.16549347]]),
'sites': {'ontop': (0, 0),
'bridge': (0.5, 0),
'fcc': (0.3333333333333333, 0.3333333333333333),
'hcp': (0.6666666666666666, 0.6666666666666666)},
'top layer atom index': 48}}
今回の例fcc111だと、 ontop, bridge, fcc, hcp があるようです。それぞれ実際に見てみましょう。
[15]:
orthogonal = False
slab_mol_ontop = hcp0001("Co", a=a, c=c, size=(4, 4, 4), vacuum=40.0, periodic=True, orthogonal=orthogonal)
mol = molecule("CO")
add_adsorbate(slab_mol_ontop, mol, 3.0, "ontop")
slab_mol_bridge = hcp0001("Co", a=a, c=c, size=(4, 4, 4), vacuum=40.0, periodic=True, orthogonal=orthogonal)
mol = molecule("CO")
add_adsorbate(slab_mol_bridge, mol, 3.0, "bridge")
slab_mol_fcc = hcp0001("Co", a=a, c=c, size=(4, 4, 4), vacuum=40.0, periodic=True, orthogonal=orthogonal)
mol = molecule("CO")
add_adsorbate(slab_mol_fcc, mol, 3.0, "fcc")
slab_mol_hcp = hcp0001("Co", a=a, c=c, size=(4, 4, 4), vacuum=40.0, periodic=True, orthogonal=orthogonal)
mol = molecule("CO")
add_adsorbate(slab_mol_hcp, mol, 3.0, "hcp")
[16]:
view_ngl([slab_mol_ontop, slab_mol_bridge, slab_mol_fcc, slab_mol_hcp])
[16]:
[17]:
from ase.io import write
from IPython.display import Image
import matplotlib.pyplot as plt
import matplotlib.image as mpimg
write("output/co_ontop.png", slab_mol_ontop, rotation="0x,0y,90z")
write("output/co_bridge.png", slab_mol_bridge, rotation="0x,0y,90z")
write("output/co_fcc.png", slab_mol_fcc, rotation="0x,0y,90z")
write("output/co_hcp.png", slab_mol_hcp, rotation="0x,0y,90z")
fig, axes = plt.subplots(1, 4, figsize=(12, 3))
for i, adsorbate_name in enumerate(["ontop", "bridge", "fcc", "hcp"]):
ax = axes[i]
ax.imshow(mpimg.imread(f"output/pt_{adsorbate_name}.png"))
ax.set_axis_off()
ax.set_title(adsorbate_name)
fig.show()
上記のように、
ontop: ある1つの原子の上bridge: ある2つの原子の間fcc: 3つの原子の間で、2層目には原子が存在しない位置hcp: 3つの原子の間で、2層目の原子の直上の位置
という形となっています。
分子が表面構造のどの部分に吸着するのか(吸着サイト)は、表面・分子系によって異なり自明ではありません。 吸着エネルギーが最小(吸着構造のエネルギーが最小)となる場所を探す必要があります。
[18]:
E_slab_mol_ontop = get_opt_energy(slab_mol_ontop, fmax=1e-3)
E_slab_mol_bridge = get_opt_energy(slab_mol_bridge, fmax=1e-3)
E_slab_mol_fcc = get_opt_energy(slab_mol_fcc, fmax=1e-3)
E_slab_mol_hcp = get_opt_energy(slab_mol_hcp, fmax=1e-3)
Step Time Energy fmax
LBFGS: 0 05:01:09 -317.842408 2.898524
LBFGS: 1 05:01:09 -317.970382 3.594473
LBFGS: 2 05:01:09 -318.191282 1.677129
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calculator)
LBFGS: 3 05:01:09 -318.281448 0.675228
LBFGS: 4 05:01:09 -318.290984 0.402372
LBFGS: 5 05:01:09 -318.298270 0.037055
LBFGS: 6 05:01:09 -318.299179 0.060686
LBFGS: 7 05:01:09 -318.302309 0.184551
LBFGS: 8 05:01:09 -318.304047 0.160590
LBFGS: 9 05:01:09 -318.304851 0.053999
LBFGS: 10 05:01:09 -318.305001 0.024050
LBFGS: 11 05:01:09 -318.305081 0.019806
LBFGS: 12 05:01:10 -318.305105 0.025294
LBFGS: 13 05:01:10 -318.305111 0.013119
LBFGS: 14 05:01:10 -318.305159 0.004592
LBFGS: 15 05:01:10 -318.305176 0.004777
LBFGS: 16 05:01:10 -318.305135 0.009718
LBFGS: 17 05:01:10 -318.305175 0.010435
LBFGS: 18 05:01:10 -318.305210 0.005958
LBFGS: 19 05:01:10 -318.305214 0.001659
LBFGS: 20 05:01:10 -318.305143 0.002806
LBFGS: 21 05:01:10 -318.305147 0.003268
LBFGS: 22 05:01:10 -318.305220 0.002370
LBFGS: 23 05:01:10 -318.305214 0.001250
LBFGS: 24 05:01:10 -318.305179 0.001596
LBFGS: 25 05:01:11 -318.305176 0.002351
LBFGS: 26 05:01:11 -318.305144 0.002218
LBFGS: 27 05:01:11 -318.305219 0.001216
LBFGS: 28 05:01:11 -318.305215 0.000552
Step Time Energy fmax
LBFGS: 0 05:01:11 -317.298018 3.367196
LBFGS: 1 05:01:11 -317.432625 4.081287
LBFGS: 2 05:01:11 -317.699279 2.858173
LBFGS: 3 05:01:11 -317.973906 1.086518
LBFGS: 4 05:01:11 -317.996374 0.707769
LBFGS: 5 05:01:11 -318.071183 0.641063
LBFGS: 6 05:01:11 -318.081374 0.366953
LBFGS: 7 05:01:11 -318.087611 0.250201
LBFGS: 8 05:01:11 -318.107593 0.845663
LBFGS: 9 05:01:11 -318.115866 0.647180
LBFGS: 10 05:01:12 -318.121201 0.170366
LBFGS: 11 05:01:12 -318.123988 0.213779
LBFGS: 12 05:01:12 -318.127898 0.427406
LBFGS: 13 05:01:12 -318.130784 0.379354
LBFGS: 14 05:01:12 -318.132467 0.151580
LBFGS: 15 05:01:12 -318.133094 0.059628
LBFGS: 16 05:01:12 -318.133756 0.175862
LBFGS: 17 05:01:12 -318.134530 0.215524
LBFGS: 18 05:01:12 -318.135066 0.130313
LBFGS: 19 05:01:12 -318.135305 0.023242
LBFGS: 20 05:01:12 -318.135427 0.049550
LBFGS: 21 05:01:12 -318.135580 0.089026
LBFGS: 22 05:01:12 -318.135706 0.078572
LBFGS: 23 05:01:12 -318.135824 0.030660
LBFGS: 24 05:01:12 -318.135793 0.010860
LBFGS: 25 05:01:12 -318.135857 0.030221
LBFGS: 26 05:01:13 -318.135911 0.038738
LBFGS: 27 05:01:13 -318.135879 0.024980
LBFGS: 28 05:01:13 -318.135890 0.007104
LBFGS: 29 05:01:13 -318.135900 0.009158
LBFGS: 30 05:01:13 -318.135897 0.017172
LBFGS: 31 05:01:13 -318.135899 0.018328
LBFGS: 32 05:01:13 -318.135899 0.010433
LBFGS: 33 05:01:13 -318.135916 0.004518
LBFGS: 34 05:01:13 -318.135891 0.010585
LBFGS: 35 05:01:13 -318.135949 0.017736
LBFGS: 36 05:01:13 -318.135855 0.019821
LBFGS: 37 05:01:13 -318.135927 0.013268
LBFGS: 38 05:01:13 -318.135985 0.005323
LBFGS: 39 05:01:13 -318.135954 0.018906
LBFGS: 40 05:01:13 -318.135961 0.037519
LBFGS: 41 05:01:13 -318.135991 0.049869
LBFGS: 42 05:01:14 -318.136023 0.043147
LBFGS: 43 05:01:14 -318.136093 0.017429
LBFGS: 44 05:01:14 -318.136120 0.015111
LBFGS: 45 05:01:14 -318.136138 0.040347
LBFGS: 46 05:01:14 -318.136259 0.055027
LBFGS: 47 05:01:14 -318.136321 0.044535
LBFGS: 48 05:01:14 -318.134209 0.175405
LBFGS: 49 05:01:14 -318.136430 0.021860
LBFGS: 50 05:01:14 -318.136463 0.012867
LBFGS: 51 05:01:14 -318.136512 0.060926
LBFGS: 52 05:01:14 -318.136521 0.025587
LBFGS: 53 05:01:14 -318.136524 0.032892
LBFGS: 54 05:01:15 -318.136518 0.029711
LBFGS: 55 05:01:15 -318.136533 0.052500
LBFGS: 56 05:01:15 -318.136558 0.055009
LBFGS: 57 05:01:15 -318.136564 0.041614
LBFGS: 58 05:01:15 -318.136645 0.025558
LBFGS: 59 05:01:15 -318.136664 0.011149
LBFGS: 60 05:01:15 -318.136694 0.028943
LBFGS: 61 05:01:15 -318.136706 0.042383
LBFGS: 62 05:01:15 -318.136710 0.028344
LBFGS: 63 05:01:15 -318.136797 0.008639
LBFGS: 64 05:01:15 -318.136819 0.019556
LBFGS: 65 05:01:15 -318.136773 0.033059
LBFGS: 66 05:01:15 -318.136823 0.034832
LBFGS: 67 05:01:16 -318.136881 0.020040
LBFGS: 68 05:01:16 -318.136843 0.005330
LBFGS: 69 05:01:16 -318.136902 0.015483
LBFGS: 70 05:01:16 -318.136883 0.022676
LBFGS: 71 05:01:16 -318.136915 0.019674
LBFGS: 72 05:01:16 -318.136884 0.008090
LBFGS: 73 05:01:16 -318.136937 0.002205
LBFGS: 74 05:01:16 -318.136921 0.006972
LBFGS: 75 05:01:16 -318.136944 0.009053
LBFGS: 76 05:01:16 -318.136934 0.006030
LBFGS: 77 05:01:16 -318.136941 0.001094
LBFGS: 78 05:01:16 -318.136933 0.001779
LBFGS: 79 05:01:17 -318.136942 0.003125
LBFGS: 80 05:01:17 -318.136944 0.002603
LBFGS: 81 05:01:17 -318.136938 0.000913
Step Time Energy fmax
LBFGS: 0 05:01:17 -317.197130 3.303990
LBFGS: 1 05:01:17 -317.316486 4.021271
LBFGS: 2 05:01:17 -317.552021 2.731868
LBFGS: 3 05:01:17 -317.811816 1.460499
LBFGS: 4 05:01:17 -317.847270 1.047521
LBFGS: 5 05:01:17 -317.982363 1.271480
LBFGS: 6 05:01:17 -318.026900 1.048226
LBFGS: 7 05:01:17 -318.057192 0.314735
LBFGS: 8 05:01:18 -318.064142 0.309280
LBFGS: 9 05:01:18 -318.086642 0.740661
LBFGS: 10 05:01:18 -318.096534 0.474637
LBFGS: 11 05:01:18 -318.100364 0.116436
LBFGS: 12 05:01:18 -318.103292 0.220766
LBFGS: 13 05:01:18 -318.106737 0.353436
LBFGS: 14 05:01:18 -318.109138 0.265440
LBFGS: 15 05:01:18 -318.110216 0.078129
LBFGS: 16 05:01:18 -318.110673 0.067056
LBFGS: 17 05:01:18 -318.111208 0.155902
LBFGS: 18 05:01:18 -318.111794 0.161744
LBFGS: 19 05:01:18 -318.112151 0.078354
LBFGS: 20 05:01:19 -318.112297 0.018218
LBFGS: 21 05:01:19 -318.112343 0.045253
LBFGS: 22 05:01:19 -318.112436 0.065510
LBFGS: 23 05:01:19 -318.112558 0.047467
LBFGS: 24 05:01:19 -318.112595 0.012059
LBFGS: 25 05:01:19 -318.112616 0.014105
LBFGS: 26 05:01:19 -318.112630 0.031005
LBFGS: 27 05:01:19 -318.112652 0.031259
LBFGS: 28 05:01:19 -318.112681 0.014552
LBFGS: 29 05:01:19 -318.112650 0.002562
LBFGS: 30 05:01:19 -318.112651 0.004874
LBFGS: 31 05:01:19 -318.112656 0.007728
LBFGS: 32 05:01:19 -318.112677 0.005742
LBFGS: 33 05:01:20 -318.112668 0.001651
LBFGS: 34 05:01:20 -318.112664 0.000739
Step Time Energy fmax
LBFGS: 0 05:01:20 -317.202676 3.329409
LBFGS: 1 05:01:20 -317.323988 4.036102
LBFGS: 2 05:01:20 -317.562339 2.758290
LBFGS: 3 05:01:20 -317.827578 1.460115
LBFGS: 4 05:01:20 -317.863367 1.048154
LBFGS: 5 05:01:20 -317.998344 1.243057
LBFGS: 6 05:01:20 -318.041644 1.015544
LBFGS: 7 05:01:20 -318.070363 0.316262
LBFGS: 8 05:01:20 -318.077359 0.315765
LBFGS: 9 05:01:20 -318.102029 0.707727
LBFGS: 10 05:01:20 -318.111213 0.437634
LBFGS: 11 05:01:20 -318.115049 0.176868
LBFGS: 12 05:01:21 -318.117735 0.215533
LBFGS: 13 05:01:21 -318.121458 0.348068
LBFGS: 14 05:01:21 -318.124152 0.271505
LBFGS: 15 05:01:21 -318.125450 0.083579
LBFGS: 16 05:01:21 -318.125985 0.055497
LBFGS: 17 05:01:21 -318.126532 0.138587
LBFGS: 18 05:01:21 -318.127129 0.141324
LBFGS: 19 05:01:21 -318.127410 0.065595
LBFGS: 20 05:01:21 -318.127459 0.015101
LBFGS: 21 05:01:21 -318.127602 0.042058
LBFGS: 22 05:01:21 -318.127698 0.063686
LBFGS: 23 05:01:21 -318.127806 0.049576
LBFGS: 24 05:01:22 -318.127834 0.014832
LBFGS: 25 05:01:22 -318.127799 0.011298
LBFGS: 26 05:01:22 -318.127835 0.025439
LBFGS: 27 05:01:22 -318.127860 0.025955
LBFGS: 28 05:01:22 -318.127912 0.012118
LBFGS: 29 05:01:22 -318.127881 0.001567
LBFGS: 30 05:01:22 -318.127888 0.003975
LBFGS: 31 05:01:22 -318.127853 0.006622
LBFGS: 32 05:01:22 -318.127849 0.005111
LBFGS: 33 05:01:22 -318.127903 0.001587
LBFGS: 34 05:01:22 -318.127830 0.000604
[19]:
import pandas as pd
series = pd.Series({
"ontop": E_slab_mol_ontop,
"bridge": E_slab_mol_bridge,
"fcc": E_slab_mol_fcc,
"hcp": E_slab_mol_hcp,
})
E_adsorp_series = series - (E_slab + E_mol)
E_adsorp_series.plot(style="o-", xlabel="Adsorption site", ylabel="Energy [eV]", title="Adsorption energy for each adsorption site")
plt.show()
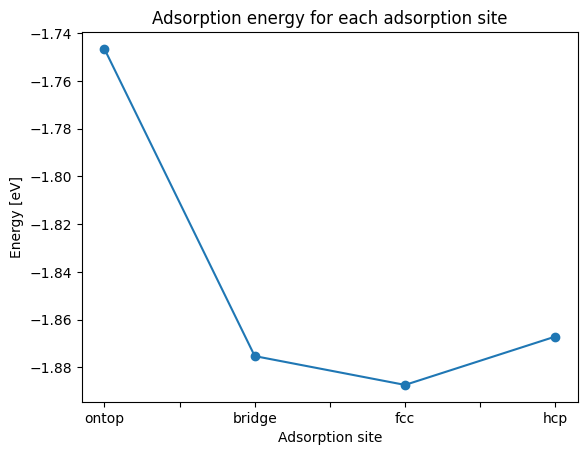
このように、吸着サイトによって吸着構造のエネルギーが異なることがわかります。 今回の計算では、ontopサイトへの吸着構造が一番安定で、吸着エネルギーが一番小さいという結果が得られました。
[20]:
view_ngl([slab_mol_ontop, slab_mol_bridge, slab_mol_fcc, slab_mol_hcp])
[20]:
FixAtoms constraints を使用する例¶
上記の計算例では、すべての原子座標を構造緩和しましたが、DFTを用いて吸着構造を求める場合、構造の安定性を高めるためにSlabの下層の原子座標を固定して構造緩和を行うことがあります。
この方法での計算も行ってみましょう。ASEのconstraintであるFixAtomsを用いることで、指定した index の原子座標を固定することができます。
[21]:
%%time
slab_mol_fixed = hcp0001("Co", a=a, c=c, size=(4, 4, 4), vacuum=40.0, periodic=True, orthogonal=orthogonal)
mol2 = molecule("CO")
add_adsorbate(slab_mol_fixed, mol2, 3.0, "bridge")
view_ngl(slab_mol_fixed)
CPU times: user 34.8 ms, sys: 8.03 ms, total: 42.8 ms
Wall time: 37.6 ms
[21]:
今回は下から2層の原子を指定する際に、z座標でしきい値を設定します。
上記の可視化結果をマウスホバーしてもわかりますが、今回は以下のようにz軸の分布を可視化して確認してみます。
[22]:
import pandas as pd
atoms = slab_mol_fixed
df = pd.DataFrame({
"x": atoms.positions[:, 0],
"y": atoms.positions[:, 1],
"z": atoms.positions[:, 2],
"symbol": atoms.symbols,
})
df
[22]:
| x | y | z | symbol | |
|---|---|---|---|---|
| 0 | -4.626848e-17 | 1.443662 | 40.00000 | Co |
| 1 | 2.500496e+00 | 1.443662 | 40.00000 | Co |
| 2 | 5.000993e+00 | 1.443662 | 40.00000 | Co |
| 3 | 7.501489e+00 | 1.443662 | 40.00000 | Co |
| 4 | 1.250248e+00 | 3.609156 | 40.00000 | Co |
| ... | ... | ... | ... | ... |
| 61 | 6.251241e+00 | 6.496480 | 46.03421 | Co |
| 62 | 8.751738e+00 | 6.496480 | 46.03421 | Co |
| 63 | 1.125223e+01 | 6.496480 | 46.03421 | Co |
| 64 | 1.250248e+00 | 0.000000 | 49.03421 | O |
| 65 | 1.250248e+00 | 0.000000 | 47.88387 | C |
66 rows × 4 columns
[23]:
import plotly.express as px
coord = "z"
df_sorted = df.sort_values(coord).reset_index().rename({"index": "atom_index"}, axis=1)
fig = px.scatter(df_sorted, x=df_sorted.index, y=coord, color="symbol", hover_data=["x", "y", "z", "atom_index"])
fig.show()
2層目と3層目の境界が、42.03 - 44.07 の間であることがわかりました。
FixAtoms は indicesに固定したい原子のindexを指定するか、maskにそれぞれの原子を固定するかしないかの配列を指定します。 今回はmaskを使用してみます。
[24]:
from ase.constraints import FixAtoms
thresh = 43.0
constraint = FixAtoms(mask=slab_mol_fixed.positions[:, 2] < thresh)
constraint
[24]:
FixAtoms(indices=[0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31])
作成したconstraintは、 atoms.set_constraint methodで指定することで制約がかけられます。
[25]:
slab_mol_fixed.set_constraint(constraint)
可視化して、意図した場所を固定できているか確認してみます。
[26]:
v = view_ngl(slab_mol_fixed)
v.show_fix_atoms_constraint()
v
[26]:
[27]:
E_slab_mol_fixed = get_opt_energy(slab_mol_fixed, fmax=1e-3)
Step Time Energy fmax
LBFGS: 0 05:01:23 -317.298046 3.367193
LBFGS: 1 05:01:23 -317.370433 4.084981
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning:
Please use atoms.calc = calc
LBFGS: 2 05:01:23 -317.549330 2.528261
LBFGS: 3 05:01:24 -317.833422 2.157216
LBFGS: 4 05:01:24 -317.865740 0.838110
LBFGS: 5 05:01:24 -317.899339 0.966453
LBFGS: 6 05:01:24 -317.922718 1.125139
LBFGS: 7 05:01:24 -317.936185 0.675126
LBFGS: 8 05:01:24 -317.943700 0.249672
LBFGS: 9 05:01:24 -317.948898 0.451393
LBFGS: 10 05:01:24 -317.956971 0.705947
LBFGS: 11 05:01:24 -317.963627 0.549626
LBFGS: 12 05:01:24 -317.966968 0.200157
LBFGS: 13 05:01:24 -317.968894 0.193649
LBFGS: 14 05:01:24 -317.971012 0.367471
LBFGS: 15 05:01:24 -317.973073 0.366748
LBFGS: 16 05:01:24 -317.974495 0.185133
LBFGS: 17 05:01:25 -317.975121 0.052330
LBFGS: 18 05:01:25 -317.975567 0.124829
LBFGS: 19 05:01:25 -317.976069 0.179915
LBFGS: 20 05:01:25 -317.976485 0.130129
LBFGS: 21 05:01:25 -317.976679 0.036992
LBFGS: 22 05:01:25 -317.976742 0.032473
LBFGS: 23 05:01:25 -317.976841 0.067961
LBFGS: 24 05:01:25 -317.976913 0.067908
LBFGS: 25 05:01:25 -317.976948 0.032542
LBFGS: 26 05:01:25 -317.976991 0.006594
LBFGS: 27 05:01:25 -317.977004 0.018813
LBFGS: 28 05:01:26 -317.977011 0.032035
LBFGS: 29 05:01:26 -317.977034 0.029605
LBFGS: 30 05:01:26 -317.977043 0.013607
LBFGS: 31 05:01:26 -317.977053 0.005401
LBFGS: 32 05:01:26 -317.977055 0.012157
LBFGS: 33 05:01:26 -317.977064 0.017967
LBFGS: 34 05:01:26 -317.977071 0.015757
LBFGS: 35 05:01:26 -317.977069 0.007217
LBFGS: 36 05:01:26 -317.977020 0.007363
LBFGS: 37 05:01:26 -317.977075 0.017915
LBFGS: 38 05:01:26 -317.977102 0.029666
LBFGS: 39 05:01:26 -317.977073 0.035775
LBFGS: 40 05:01:27 -317.977122 0.027862
LBFGS: 41 05:01:27 -317.977149 0.010232
LBFGS: 42 05:01:27 -317.977187 0.019037
LBFGS: 43 05:01:27 -317.977211 0.042417
LBFGS: 44 05:01:27 -317.977241 0.055570
LBFGS: 45 05:01:27 -317.977295 0.044879
LBFGS: 46 05:01:27 -317.977276 0.015693
LBFGS: 47 05:01:27 -317.977347 0.011269
LBFGS: 48 05:01:27 -317.977329 0.028472
LBFGS: 49 05:01:27 -317.977419 0.037660
LBFGS: 50 05:01:27 -317.976700 0.042962
LBFGS: 51 05:01:28 -317.977447 0.028700
LBFGS: 52 05:01:28 -317.977466 0.020285
LBFGS: 53 05:01:28 -317.975169 0.217418
LBFGS: 54 05:01:28 -317.977530 0.010444
LBFGS: 55 05:01:28 -317.977554 0.010728
LBFGS: 56 05:01:28 -317.976147 0.167147
LBFGS: 57 05:01:28 -317.977605 0.015344
LBFGS: 58 05:01:28 -317.977641 0.018246
LBFGS: 59 05:01:28 -317.975360 0.152353
LBFGS: 60 05:01:28 -317.977710 0.019435
LBFGS: 61 05:01:29 -317.977729 0.020207
LBFGS: 62 05:01:29 -317.977740 0.019599
LBFGS: 63 05:01:29 -317.977792 0.015333
LBFGS: 64 05:01:29 -317.977923 0.026060
LBFGS: 65 05:01:29 -317.977937 0.029808
LBFGS: 66 05:01:29 -317.978010 0.027831
LBFGS: 67 05:01:29 -317.978059 0.012066
LBFGS: 68 05:01:29 -317.978089 0.013912
LBFGS: 69 05:01:29 -317.978113 0.022272
LBFGS: 70 05:01:29 -317.978131 0.025347
LBFGS: 71 05:01:30 -317.978144 0.016545
LBFGS: 72 05:01:30 -317.978148 0.002875
LBFGS: 73 05:01:30 -317.978164 0.004765
LBFGS: 74 05:01:30 -317.978164 0.007394
LBFGS: 75 05:01:30 -317.978161 0.006962
LBFGS: 76 05:01:30 -317.978175 0.003307
LBFGS: 77 05:01:30 -317.978126 0.001155
LBFGS: 78 05:01:30 -317.978161 0.002111
LBFGS: 79 05:01:30 -317.978178 0.003076
LBFGS: 80 05:01:31 -317.978163 0.002670
LBFGS: 81 05:01:31 -317.978175 0.001194
LBFGS: 82 05:01:31 -317.978169 0.000279
[28]:
E_adsorp = E_slab_mol_fixed - (E_slab + E_mol)
print(f"E_adsorp {E_adsorp}, E_slab_mol {E_slab_mol_fixed}, E_slab {E_slab}, E_mol {E_mol}")
E_adsorp -1.3960754815630594, E_slab_mol -317.9781686732514, E_slab -305.13616061153743, E_mol -11.445932580150906
下層を固定する方法を用いた場合、吸着エネルギーとして-1.19eVが得られました。
CO on Pd¶
同様に、Pd上でのCO吸着に関しても計算してみましょう。
[29]:
import numpy as np
bulk_atoms = bulk("Pd", cubic=True)
np.mean(np.diag(bulk_atoms.cell))
[29]:
3.89
[30]:
bulk_atoms.calc = calculator
E_bulk = get_opt_energy(bulk_atoms, fmax=1e-4, opt_mode="scale")
a_pd = np.mean(np.diag(bulk_atoms.cell))
a_pd
Step Time Energy fmax
LBFGS: 0 05:01:31 -14.904795 4.593262
LBFGS: 1 05:01:31 -14.805114 7.050895
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning:
Please use atoms.calc = calc
/tmp/ipykernel_3433/1344539460.py:9: FutureWarning:
Import StrainFilter from ase.filters
LBFGS: 2 05:01:31 -14.954530 0.571581
LBFGS: 3 05:01:31 -14.955401 0.069699
LBFGS: 4 05:01:31 -14.955413 0.000910
LBFGS: 5 05:01:31 -14.955418 0.000067
[30]:
3.941105705982878
[31]:
from ase.build import fcc111, molecule, add_adsorbate
slab = fcc111("Pd", a=a_pd, size=(4, 4, 4), vacuum=40.0, periodic=True)
slab.calc = calculator
E_slab = get_opt_energy(slab, fmax=1e-4, opt_mode="normal")
Step Time Energy fmax
LBFGS: 0 05:01:31 -221.469662 0.154306
LBFGS: 1 05:01:31 -221.479485 0.123494
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning:
Please use atoms.calc = calc
LBFGS: 2 05:01:32 -221.497979 0.037257
LBFGS: 3 05:01:32 -221.498799 0.037970
LBFGS: 4 05:01:32 -221.502356 0.031879
LBFGS: 5 05:01:32 -221.504820 0.024933
LBFGS: 6 05:01:32 -221.505937 0.009735
LBFGS: 7 05:01:32 -221.506049 0.001465
LBFGS: 8 05:01:32 -221.505994 0.000064
[32]:
mol = molecule("CO")
mol.calc = calculator
E_mol = get_opt_energy(mol, fmax=1e-4)
Step Time Energy fmax
LBFGS: 0 05:01:32 -11.443040 0.797118
LBFGS: 1 05:01:32 -11.431572 1.878737
LBFGS: 2 05:01:32 -11.445922 0.046010
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning:
Please use atoms.calc = calc
LBFGS: 3 05:01:32 -11.445935 0.002584
LBFGS: 4 05:01:32 -11.445933 0.000005
[33]:
%%time
slab_mol = fcc111("Pd", a=a_pd, size=(4, 4, 4), vacuum=40.0)
slab_mol.pbc = True
mol2 = molecule("CO")
add_adsorbate(slab_mol, mol2, 3.0, "ontop")
E_slab_mol = get_opt_energy(slab_mol, fmax=1e-3)
Step Time Energy fmax
LBFGS: 0 05:01:32 -234.320127 0.953715
LBFGS: 1 05:01:32 -234.323178 1.770322
/tmp/ipykernel_3433/1344539460.py:7: FutureWarning:
Please use atoms.calc = calc
LBFGS: 2 05:01:33 -234.347470 0.538091
LBFGS: 3 05:01:33 -234.365402 0.473518
LBFGS: 4 05:01:33 -234.371691 0.360437
LBFGS: 5 05:01:33 -234.374841 0.099046
LBFGS: 6 05:01:33 -234.378273 0.241564
LBFGS: 7 05:01:33 -234.382273 0.348405
LBFGS: 8 05:01:33 -234.384511 0.231276
LBFGS: 9 05:01:33 -234.385570 0.061829
LBFGS: 10 05:01:33 -234.386380 0.145362
LBFGS: 11 05:01:33 -234.387563 0.253167
LBFGS: 12 05:01:33 -234.388719 0.241592
LBFGS: 13 05:01:34 -234.389416 0.121320
LBFGS: 14 05:01:34 -234.389756 0.048352
LBFGS: 15 05:01:34 -234.390049 0.111616
LBFGS: 16 05:01:34 -234.390455 0.144710
LBFGS: 17 05:01:34 -234.390915 0.120400
LBFGS: 18 05:01:34 -234.391242 0.046067
LBFGS: 19 05:01:34 -234.391444 0.028925
LBFGS: 20 05:01:34 -234.391634 0.079809
LBFGS: 21 05:01:34 -234.391843 0.091986
LBFGS: 22 05:01:34 -234.391979 0.054356
LBFGS: 23 05:01:34 -234.392042 0.017186
LBFGS: 24 05:01:34 -234.392074 0.017494
LBFGS: 25 05:01:34 -234.392081 0.022621
LBFGS: 26 05:01:35 -234.392087 0.014591
LBFGS: 27 05:01:35 -234.392116 0.007934
LBFGS: 28 05:01:35 -234.392105 0.009305
LBFGS: 29 05:01:35 -234.392121 0.005786
LBFGS: 30 05:01:35 -234.392116 0.005674
LBFGS: 31 05:01:35 -234.392125 0.004706
LBFGS: 32 05:01:35 -234.392135 0.004091
LBFGS: 33 05:01:35 -234.392113 0.003857
LBFGS: 34 05:01:35 -234.392124 0.003134
LBFGS: 35 05:01:35 -234.392117 0.004607
LBFGS: 36 05:01:35 -234.392149 0.004598
LBFGS: 37 05:01:36 -234.392152 0.002170
LBFGS: 38 05:01:36 -234.392129 0.000932
CPU times: user 221 ms, sys: 18.1 ms, total: 239 ms
Wall time: 3.27 s
[34]:
E_adsorp = E_slab_mol - (E_slab + E_mol)
print(f"E_adsorp {E_adsorp}, E_slab_mol {E_slab_mol}, E_slab {E_slab}, E_mol {E_mol}")
E_adsorp -1.4402008748295714, E_slab_mol -234.39212860995127, E_slab -221.50599434099485, E_mol -11.445933394126861
[35]:
from pfcc_extras.visualize.view import view_ngl
view_ngl(slab_mol)
[35]:
このように、様々な表面や分子の組み合わせに対して吸着エネルギーが計算できます。 これは5章で説明する、効率の良い触媒探索に際して欠かせないプロセスとなります。
被覆率¶
本Tutorialで取り上げた例では、まっさらな表面構造上に、分子が1つだけ吸着するというものでした。 現実では、たくさんの分子が同時に表面と反応を起こし、同時に複数の分子が吸着しているような場合や、ほとんど吸着した分子で覆われているような場合も存在します。
その際には、分子がすでに複数個吸着している表面構造から、あらたに1つの分子が吸着する場合の吸着エネルギーなどにも興味が出てきます。
上記で上げたPd表面上でのCO分子の吸着エネルギーの被覆率依存性の解析に関しては以下の文献で報告されています。
複数箇所への吸着¶
また、今回扱えていない応用トピックとして、2箇所以上に吸着する例があります。 分子が大きく、複雑になってくると複数箇所が表面上に吸着することで安定となるようなものも存在します。